



C’est l’histoire d’un syndrome dont la cause n’a été découverte qu’en 2024 par deux équipes indépendantes de biologistes moléculaires. Cette avancée met en lumière le rôle pathologique que peuvent jouer de minuscules ARN dits non codants, dont la quantité et l’importance fonctionnelle ont longtemps été sous-estimées.
Comme chacun sait, l’ADN qui compose notre génome est copié en ARN messager lors d’une étape appelée transcription. Cet ARN transcrit est ensuite traduit en protéines. C’est l’étape de la traduction. Grâce au séquençage et aux progrès de l’analyse bio-informatique, les biologistes moléculaires se sont rapidement aperçus qu’un grand nombre d’ARN transcrits ne codent pas de protéines. Il s’agit d’ARN dits non codants.
Aujourd’hui, les tests génétiques cliniques ciblent presque uniquement les gènes codant des protéines, là où se situent la plupart des mutations responsables de maladies monogéniques, ces maladies dues à l’altération d’un seul gène. Mais avec l’essor des grands programmes de séquençage, et l’utilisation de plus en plus fréquente du séquençage du génome entier par rapport à l’exome (qui ne cible que les parties codantes), le rôle des régions non codantes dans l’apparition de maladies apparaît de plus en plus clairement.
Des travaux récents ont montré que des variations génétiques situées dans ces régions non codantes pouvaient, elles aussi, provoquer des troubles du neuro-développement. Ces altérations agissent via différents mécanismes, notamment en perturbant l’expression ou la fonction de certains ARN non codants qui, comme déjà dit, contrairement aux ARN messagers, ne servent pas à fabriquer des protéines.
Loin d’être inactifs, les ARN non codants participent à de nombreux processus cellulaires essentiels. Leur dérégulation joue un rôle dans de nombreuses maladies humaines, allant des cancers à certaines affections neurologiques.
Pour bien comprendre, il faut savoir que les parties d’un gène qui servent à fabriquer une protéine sont appelées exons, tandis que les segments non codants sont appelés introns.
Épissage : excision des introns et ligature des exons
Lors de la transcription de l’ADN, la cellule produit d’abord un ARN pré-messager, sorte de brouillon qui contient à la fois des exons et des introns. Avant de pouvoir être traduit en protéine, ce précurseur subit une étape de maturation à l’intérieur du noyau cellulaire.
C’est là qu’intervient un processus clé : l’épissage. Les introns sont retirés (excisés) du brouillon, puis les exons restants sont assemblés (ligaturés) les uns aux autres. Il en résulte un ARN messager mature, qui ne contient plus que les instructions essentielles à la fabrication d’une protéine. Cette étape garantit que l’information génétique sera correctement transmise et utilisable par la cellule.
Epissage alternatif : créer de la diversité protéique à partir d’un seul gène
Un même gène peut toutefois donner naissance à plusieurs versions d’ARN messager, et donc à plusieurs protéines, grâce à un mécanisme plus élaboré : l’épissage alternatif. Grâce à ce mécanisme, différentes combinaisons d’exons peuvent être assemblées selon le type de cellule. Un exon peut ainsi être inclus dans certains ARN messagers et exclu dans d’autres, ce qui génère une grande diversité d’ARN messagers à partir d’un seul gène. Par exemple, un gène comportant huit exons pourrait produire une première protéine à partir des exons 1, 3 et 7, une deuxième à partir des exons 2, 4, 6 et 8, et une troisième à partir des exons 1, 4, 5 et 7. Trois protéines différentes issues d’un même gène !
Cette plasticité explique comment les quelque 20 000 à 25 000 gènes humains peuvent coder pour plus de 100 000 protéines aux fonctions variées dans tous les tissus et organes. En modulant la composition des ARN messagers, l’épissage alternatif constitue une formidable stratégie évolutive pour accroître la diversité du vivant à partir d’un nombre limité de gènes. L’épissage alternatif permet de produire plusieurs ARN messagers matures à partir d’un même ARN pré-messager, en particulier dans le cerveau.
Le splicéosome assure l’excision des introns et la ligature des exons
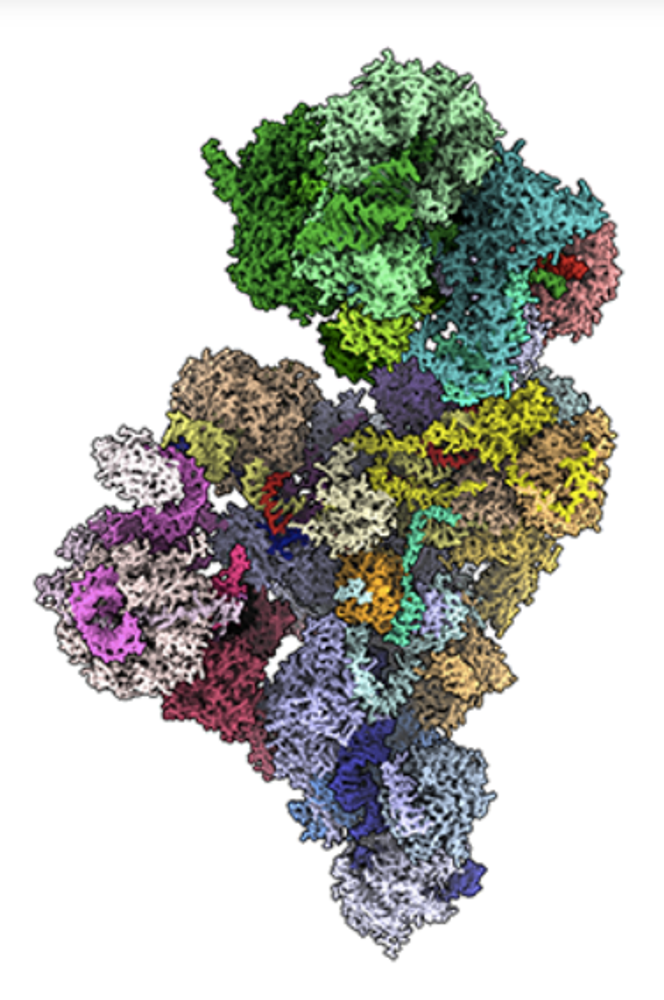
Le splicéosome – dont le nom vient du verbe anglais to splice, « épisser », c’est-à-dire assembler ou joindre – est chargé de réaliser des opérations complexes : il excise les introns et rapproche les exons, guidant ainsi la découpe précise des introns suivie de la ligature des exons. Ce processus permet la maturation correcte de l’ARN messager dans le noyau de la cellule. La réussite de ces opérations repose sur la reconnaissance de motifs spécifiques présents dans la séquence de l’ARN pré-messager.
On distingue deux types de splicéosome, au fonctionnement similaire : le splicéosome majeur et le splicéosome mineur, chacun reconnaissant des séquences génétiques distinctes. À noter que moins de 0,5 % des introns sont pris en charge (épissés) par le splicéosome mineur.
Le splicéosome majeur est une machinerie complexe composée d’environ 100 protéines et de cinq familles de petits ARN nucléaires (snRNA, small nuclear RNA) – U1, U2, U4, U5 et U6 – chacun codé par plusieurs gènes. À noter qu’il n’existe pas de U3 dans le splicéosome majeur : cet ARN est non impliqué dans l’épissage. Il joue un rôle différent et essentiel dans la maturation des ARN ribosomiques, un autre type d’ARN.
Dans le noyau de la cellule, les snRNA et les protéines s’associent pour former des particules appelées snRNP (small nuclear ribonucleoproteins), qui constituent le cœur du processus d’épissage. Chaque snRNA possède des motifs de séquence qui servent de balises, ainsi qu’une structure complexe en tiges (stems) et boucles (loops). Ces caractéristiques lui permettent d’interagir avec l’ARN pré-messager avec une précision remarquable, au bon endroit et au bon moment.
Cette mécanique sophistiquée repose sur une coopération parfaite entre les petits ARN nucléaires et les protéines du splicéosome, aboutissant à la production d’un ARN messager mature, dépourvu d’introns, prêt à être exporté dans le cytoplasme pour la traduction en protéine. C’est justement un gène codant pour un de ces petits ARN, dénommé RNU4-2, qui s’est récemment retrouvé au centre d’une découverte inattendue.
À vrai dire, RNU4-2 n’est pas le premier petit ARN nucléaire (snRNA) à être impliqué dans une maladie humaine. D’autres composants du splicéosome mineur, RNU12 et RNU4ATAC, sont déjà connus pour provoquer des troubles récessifs rares, la maladie ne survenant que lorsque les deux copies du gène sont altérées. Par exemple, des variantes de RNU12 provoquent un développement anormal du cervelet (ataxie cérébelleuse précoce), tandis que des variants de RNU4ATAC entraînent un syndrome congénital complexe, avec microcéphalie (petite tête), retard de croissance et de développement cognitif.
Mutations d’un gène codant… un petit ARN non codant
Revenons au syndrome RNU4-2. Tout commence presque en parallèle, de part et d’autre de l’Atlantique. À Oxford, dans les laboratoires du Big Data Institute et du Wellcome Centre for Human Genetics, Yuyang Chen et Nicola Whiffin analysent les données de près de 9 000 patients souffrant de troubles du neuro-développement inexpliqués, issus du vaste programme britannique 100 000 Genomes Project de Genomics England. Au même moment, à New York, Daniel Greene et Ernest Turro, à la tête d’une équipe du Mount Sinai, épluchent un jeu de données large et comparable. Les deux groupes ignorent alors qu’ils vont mettre au jour une anomalie nichée au cœur d’un gène quasiment inconnu, RNU4-2, responsable d’un nouveau syndrome. Ces découvertes seront publiées dans les revues Nature et Nature Medecine en mai et août 2024.
Situé sur le bras long du chromosome 12, le gène RNU4-2 ne code pas pour une protéine, contrairement à la majorité des gènes étudiés en clinique, mais produit une courte molécule d’ARN, appelée U4 small nuclear RNA (U4 snRNA), qui fait partie de la machinerie moléculaire chargée de traiter les messages génétiques dans nos cellules : le splicéosome.
On pourrait dire que RNU4-2 fabrique une petite pièce d’horlogerie indispensable au bon fonctionnement du splicéosome. Cette minuscule molécule d’ARN non codant joue en effet un rôle essentiel dans l’édition du message génétique.
Quand une minuscule insertion dans l’ADN bouleverse la machinerie d’épissage
En examinant les génomes de milliers de patients atteints d’un trouble neuro-développemental, les deux équipes repèrent une anomalie dans le gène RNU4-2. Celle-ci consiste en l’insertion d’une seule base – un simple « T » -. Cette mutation est hétérozygote, n’affectant qu’une des deux copies du gène.
Ce minuscule décalage, noté n.64_65insT, est apparu de novo chez l’enfant, c’est-à-dire spontanément, sans être hérité des parents. Il est retrouvé chez des dizaines de patients présentant déficience intellectuelle, retard moteur, hypotonie, microcéphalie, crises d’épilepsie, parfois une petite taille. Malgré son apparence anodine, cette modification mène à des tableaux cliniques remarquablement similaires.

Fait intriguant, l’insertion d’une base supplémentaire se situe dans une minuscule portion du gène : une région critique de 18 bases, où se forme un duplex : une structure double brin entre deux petits ARN partenaires, U4 et U6. Ce duo moléculaire fonctionne comme un couple inséparable au sein du splicéosome.
Cette insertion d’une seule base survient dans une région où l’ARN se replie de manière complexe, formant un « nœud » essentiel à la stabilité du couple U4-U6. Elle pourrait perturber leur liaison et perturber la reconnaissance des « sites d’épissage » (coupures-ligatures) nécessaires à la maturation correcte de l’ARN messager, une étape indispensable à la synthèse fidèle des protéines.
Une telle perturbation n’est pas sans effet, d’autant que RNU4-2 est très fortement exprimé dans le cerveau humain. Ceci explique pourquoi de minuscules altérations de ce gène peuvent avoir de telles conséquences sur le développement intellectuel et moteur.
C’est de cette relation intime entre RNU4-2 (le gène codant U4) et son partenaire U6 qu’est né le nom du syndrome : ReNU, pour RNU4-2 – related Neurodevelopmental disorder. Le nom ReNU n’a pas été choisi uniquement pour sa signification scientifique : les patients et leurs familles l’ont adopté, le prononçant renew. Ce nom symbolise le renouveau de l’espoir d’un avenir meilleur. Une manière touchante de rappeler que derrière chaque variation génétique, il y a des histoires humaines.
Pour ces familles, cette découverte met fin à des années d’incertitude sur l’origine de la maladie de leur enfant. Avant l’adoption du nom ReNU, le syndrome était connu sous le nom « NEDHAFA », acronyme de neurodevelopmental disorder with hypotonia, brain anomalies, distinctive facies, and absent language (trouble du développement neurologique avec hypotonie, anomalies cérébrales, visage particulier et absence de langage).
Les premiers chiffres ont vite donné la mesure de la découverte. Selon les deux premières études britannique et américaine, environ 0,4 % des patients présentant un trouble du neuro-développement – soit potentiellement des milliers d’individus dans le monde – pourraient être porteurs de cette anomalie. Autrement dit, ReNU apparaît comme l’une des causes les plus courantes de troubles monogéniques du neuro-développement, et désormais comme l’une des origines les plus fréquentes de déficience intellectuelle liée à l’anomalie d’un seul gène.
Les analyses ont aussi révélé un détail troublant : toutes les mutations identifiées concernaient l’allèle maternel, c’est-à-dire la copie du gène transmise par la mère. Il est peu probable que ce soit le fruit du hasard. Les chercheurs avancent plusieurs pistes : les mutations portées par les spermatozoïdes seraient peut-être éliminées avant la fécondation, ou bien le gène RNU4-2 pourrait être soumis à un phénomène d’empreinte génomique, n’exprimant que l’allèle d’origine maternelle. Certains suggèrent même qu’une mutation transmise par le père serait incompatible avec la vie embryonnaire.
Quand le spicéosome s’enraye
Des études récentes ont montré que certains variants pathogènes du gène RNU4-2 altèrent la structure tridimensionnelle normale de l’ARN U4, en modifiant notamment certaines parties en forme de tige (stems), indispensables à sa stabilité et à ses interactions avec U6.
Résultat : toute la machinerie perd sa synchronisation. Et lorsque le splicéosome fonctionne mal, l’épissage de centaines, voire de milliers de gènes peut être perturbé : les exons sont parfois mal assemblés, ou certains introns restent inclus.
Ces désorganisations du processus d’épissage peuvent également engendrer des autres anomalies moléculaires, comme la formation anormale de structures appelées R-loops. Ces boucles se forment lors de la transcription de l’ADN en ARN : le brin d’ARN nouvellement créé s’associe à son brin d’ADN complémentaire, ce qui laisse l’autre brin d’ADN momentanément isolé. Ces structures, normalement présentes de façon transitoire dans les cellules, deviennent problématiques si elles s’accumulent : elles peuvent perturber la lecture du message génétique, provoquer des cassures de l’ADN et fragiliser la stabilité du génome.
Dans le syndrome ReNU, les mutations du gène RNU4-2 semblent favoriser la formation excessive de R-loops, sans doute à cause d’un épissage défectueux des ARN. Cette accumulation pourrait contribuer aux troubles du développement cérébral observés chez ces enfants.
Des répercussions considérables sur le neuro-développement
Ce qui frappe, c’est le contraste entre l’infime taille de ces anomalies moléculaires – parfois une unique lettre ajoutée dans l’ADN d’un gène codant un ARN, qui plus est non codant – et l’ampleur de ses répercussions sur le neuro-développement.
Or le cerveau est particulièrement sensible à ce type de dérèglement. l faut savoir que le neuro-développement est un processus finement orchestré, au cours duquel l’expression des gènes intervient dans des tissus précis et à des moments particuliers tout au long du développement. Ce processus est notamment contrôlé par l’épissage alternatif, qui permet la mise en place d’un réseau neuronal correctement connecté et fonctionnel. De légères modifications de l’expression des gènes peuvent suffire à perturber cette architecture. À chaque étape du neuro-développement correspond un profil unique d’épissage alternatif. Toute perturbation de ce mécanisme peut entraîner des troubles du neuro-développement. Sur le plan moléculaire, bien des inconnues demeurent quant aux mécanismes reliant les erreurs d’épissage aux altérations structurales et fonctionnelles des tissus concernés.
Récemment, on a désigné sous le terme de « splicéosomopathies » l’ensemble de maladies causées par des variants pathogènes des gènes codant les ARN ou les protéines du splicéosome, cette machinerie qui assemble les messages génétiques avant leur traduction en protéines. Sur le plan clinique, ces pathologies affectent plus particulièrement certains systèmes, notamment le squelette craniofacial, la rétine, les membres, la moelle osseuse et la moelle épinière, ce qui se traduit par des affections telles que la rétinite pigmentaire (conduisant à une cécité progressive), l’atrophie musculaire spinale (maladie neurodégénérative) et d’autres atteintes hématologiques, musculaires, neurologiques.
Le syndrome ReNU : une des causes les plus fréquentes de déficience intellectuelle
Selon certaines estimations, environ un enfant sur 30 000 présente une déficience intellectuelle causée par des variants pathogènes du gène RNU4-2.
Aujourd’hui, le syndrome ReNU est considéré comme l’une des causes monogéniques les plus fréquentes de déficience intellectuelle. Sa fréquence se rapproche de celle du syndrome de Rett, une maladie liée à une mutation sur le chromosome X, qui provoque une régression rapide des acquisitions après quelques mois de développement normal. En Europe, ce syndrome touche environ une fille sur 10 000 à 15 000, soit 25 à 50 nouveaux cas par an en France et près de 9 000 dans le monde. Le syndrome ReNU reste toutefois moins fréquent que le syndrome du X fragile, la maladie génétique la plus répandue à l’origine d’un handicap intellectuel, de troubles du comportement et de particularités physiques (environ 1 garçon sur 5 000 et 1 fille sur 9 000).
En juin 2025, l’identification du gène RNU4-2 impliqué dans les troubles du développement intellectuel avait déjà permis à 145 familles en France d’obtenir une explication génétique aux symptômes de leur enfant. Ce nombre augmente rapidement, à mesure que le test est intégré aux diagnostics cliniques.
Diversité des manifestations cliniques liées au gène RNU4-2
Les patients atteints du syndrome ReNU présentent généralement des traits caractéristiques du visage, réunis sous le terme de dysmorphie faciale. Il s’agit notamment d’une grande bouche, d’un philtrum court (la petite dépression verticale entre le nez et la lèvre supérieure), de commissures des lèvres tournées vers le bas, de lèvres épaisses, d’yeux enfoncés, de sourcils clairsemés et parfois de strabisme. Chez les personnes plus âgées, une asymétrie du visage peut également être observée.
Si la mise en évidence du gène RNU4-2 a permis d’expliquer de nombreux cas de déficience intellectuelle jusque-là restés mystérieux, une question demeure : pourquoi certains enfants présentent-ils une forme très sévère du syndrome, tandis que d’autres sont bien moins atteints ?
C’est précisément ce qu’a cherché à comprendre une vaste étude internationale, qui s’est attachée à explorer la diversité des manifestations cliniques liées à ce gène. Publiée dans la revue Nature Genetics, elle a porté sur 23 649 patients atteints de troubles rares, ainsi que sur d’autres cas issus de collaborations internationales. Les chercheurs ont mis en évidence des variations pathogènes dans les gènes RNU4-2, mais aussi RNU5A-1 et RNU5B-1 : trois gènes impliqués dans la même machinerie cellulaire, dont les altérations entraînent des troubles du neuro-développement d’intensité variable.
Chez les patients porteurs d’un variant du gène RNU4-2, la sévérité des symptômes dépend étroitement de la région du gène touchée. Les formes les plus graves sont associées à une mutation bien précise : l’insertion d’une thymine (T) entre les positions 64 et 65 de la séquence du gène.
Ces enfants présentent souvent, dès la naissance, une hypotonie (faible tonus musculaire) et des difficultés à s’alimenter. La plupart souffrent d’un retard global du développement, marchant très tardivement (en moyenne vers 2 ans et demi) ou pas du tout (dans 13 % des cas). Le langage est très limité, parfois absent, et la déficience intellectuelle généralement sévère. Les crises d’épilepsie sont fréquentes : la majorité répond aux traitements, mais certaines formes se montrent résistantes. Plus d’un enfant sur trois présente également des anomalies squelettiques, particulièrement une ostéopénie (diminution de la densité osseuse) ou des fractures.
Les examens d’imagerie cérébrale révèlent presque toujours des anomalies, telles qu’un amincissement du corps calleux – la structure reliant les deux hémisphères cérébraux – et une dilatation des ventricules. D’autres signes peuvent accompagner le tableau clinique : petite taille, microcéphalie, traits du visage particuliers, strabisme, troubles digestifs, anomalies osseuses.
Chez les patients porteurs d’une autre mutation, n.76C > ; T (remplacement d’une cytosine par une thymine à la position 76), le tableau est nettement plus modéré : ces enfants marchent, souvent à un âge normal, possèdent de bonnes capacités langagières et leur développement intellectuel est moins altéré. Une microcéphalie et une petite taille sont plus rares.
Les mutations situées dans une autre région du gène, dite « en tige » (stem III), entraînent elles aussi des formes plus légères, avec un retard du développement variable, une déficience intellectuelle modérée et une acquisition du langage généralement conservée.
Signature épigénétique
Des chercheurs ont récemment mis en évidence, dans les lymphocytes (cellules sanguines), une signature épigénétique particulière chez les patients atteints du syndrome ReNU. Concrètement, il s’agit d’anomalies dans le profil de méthylation de l’ADN, un marqueur qui influence l’expression des gènes, et qui distingue nettement les patients des sujets témoins.
Ces altérations sont particulièrement prononcées dans les formes sévères du syndrome, notamment lorsque la mutation touche la boucle T (T-loop) : une région où l’ARN non codant se replie en une structure complexe en forme de T au sein du splicéosome.
Parallèlement, les chercheurs ont observé une signature transcriptomique, c’est-à-dire un profil caractéristique des ARN messagers produits, qui reflète l’impact de la mutation sur l’expression des gènes.
Ces découvertes ont permis de développer deux types de tests, transcriptomique et épigénétique, qui facilitent le diagnostic du syndrome ReNU. Pour de nombreuses familles, ils apportent enfin une explication tant attendue, mettant fin à des années d’incertitude et d’errance diagnostique. Ils offrent aussi un outil précieux pour le conseil génétique, en permettant aux parents de mieux évaluer le risque que d’autres enfants soient touchés par la même maladie.
Mieux comprendre le rôle pathogène des ARN non codants en neurologie
Le syndrome ReNU ne se limite pas à l’identification d’une nouvelle maladie : il marque une avancée majeure dans la compréhension du rôle des ARN non codants. Longtemps négligés, ces petits ARN se révèlent indispensables au fonctionnement du génome et, lorsqu’ils dysfonctionnent, peuvent provoquer des troubles du neuro-développement étonnamment fréquents.
Au-delà du syndrome ReNU, cette découverte ouvre une perspective inédite sur la génétique des troubles du neuro-développement, un ensemble de maladies très diverses comprenant le trouble du déficit de l’attention avec ou sans hyperactivité (TDAH), l’autisme, la déficience intellectuelle, ainsi que certains troubles spécifiques des apprentissages ou des mouvements. Aujourd’hui, on estime que 40 % à 60 % des personnes atteintes de troubles neuro-développementaux rares restent sans diagnostic malgré des bilans génétiques complets.
Les formes récessives : nouvelles facettes des syndromes liés aux gènes RNU4-2 et RNU2-2
En août 2025, deux équipes internationales ont annoncé sur la plateforme de preprints medRxiv la découverte d’une nouvelle forme de maladie liée au gène RNU4-2. Jusqu’ici, ce gène n’était associé qu’au syndrome ReNU, une maladie à transmission dominante : une seule copie altérée suffit à provoquer la maladie. Cette fois, les chercheurs ont identifié chez plusieurs enfants des anomalies dites « bialléliques », présentes sur les deux copies du gène, héritées respectivement de la mère et du père. Ces variantes de type « perte de fonction » rendent le gène totalement inactif.
Contrairement à la forme dominante, cette nouvelle forme récessive se manifeste par des symptômes neurologiques distincts, notamment des anomalies spécifiques de la substance blanche du cerveau visibles à l’imagerie. L’analyse génétique confirme que c’est la combinaison de deux copies altérées qui déclenche la maladie, distinguant clairement cette forme de la version dominante du syndrome ReNU.
Ces travaux montrent que RNU4-2 peut être à l’origine de plusieurs maladies différentes selon la nature et la localisation des anomalies génétiques. Ils élargissent ainsi à la fois le spectre des mutations et la diversité des symptômes associés aux troubles du neuro-développement liés à ce gène.
Dans la continuité de ces découvertes, d’autres gènes du splicéosome majeur ont été à leur tour mise en cause dans des formes voisines de troubles neuro-développementaux. Parmi eux, RNU2-2 et RNU5B-1, codant de petits ARN nucléaires non codants, ont été identifiés comme responsables de troubles du développement intellectuel.
RNU2-2 et RNU5B-1, nouveaux gènes responsables de troubles du développement intellectuel
En mai 2025, une étude publiée dans Nature Genetics a montré que pour RNU2-2, on observe à la fois une forme dominante et une forme récessive de la maladie, cette dernière étant même plus fréquente en Angleterre que la forme dominante. Les chercheurs précisent que, si la forme dominante du syndrome ReNU reste la plus souvent diagnostiquée, la forme récessive liée à RNU2-2 n’est pas rare : pour 10 enfants touchés par le syndrome ReNU, on compte environ 2 à 4 enfants atteints du syndrome récessif NDD lié à RNU2-2.
Une étude britannique et américaine publiée en août 2025 sur medRxiv a confirmé que, même dans des familles sans consanguinité, la forme récessive du syndrome associée à RNU2-2 est plus fréquente que la forme dominante. Les chercheurs insistent d’ailleurs sur l’importance du conseil génétique : comme il s’agit d’une maladie héréditaire, il est possible d’informer les parents avant une nouvelle grossesse ou d’envisager un diagnostic prénatal.
En septembre 2025, une équipe menée par Christel Depienne (université d’Essen, Allemagne), Caroline Nava (Institut du Cerveau, Inserm/Sorbonne Université/CNRS) et Gaétan Lesca (CHU de Lyon) a également montré que les formes récessives dues au gène RNU2-2 sont au moins deux fois plus fréquentes que les formes dominantes.
Ce travail souligne le rôle central, longtemps sous-estimé, des petits ARN non codants dans les troubles du développement d’origine génétique. Les anomalies du gène RNU2-2 sont presque aussi fréquentes que celles du gène RNU4-2, responsable du syndrome ReNU. Cliniquement, les patients présentent un trouble neuro-développemental souvent associé à une déficience intellectuelle sévère, l’épilepsie survenant dans la majorité des cas avant l’âge de 3 ans.
RNU5B-1 constitue un troisième gène codant un petit ARN nucléaire non codant, élément essentiel du complexe majeur d’épissage. Ce gène est aussi impliqué dans les troubles du neuro-développement, mais avec un profil clinique différent : la plupart des enfants présentent un retard sévère (ou parfois modéré) du développement et des anomalies cérébrales visibles à l’IRM. Certains présentent un thorax en entonnoir (pectus excavatum), un aspect longiligne (membres fins et allongés), ou des atteintes oculaires telles qu’un glaucome congénital ou une forte myopie.
En juin 2025, 18 patients atteints de troubles du neuro-développement liés au gène RNU5B-1 avaient déjà été identifiés en France.
La découverte de variants pathogènes dans les gènes RNU4-2, RNU2-2 et RNU5B-1 illustre à quel point les ARN non codants sont au cœur de la régulation génétique et combien il reste à découvrir sur les dysfonctionnements de cette machinerie dans le neuro-développement.
Mettre fin à l’errance diagnostique pour de nombreux patients
Ces travaux, le plus souvent issus de collaborations internationales, mettent en lumière l’impact direct pour les familles. Alors que la plupart des analyses génétiques classiques se concentrent sur l’ADN codant (plus de 1 500 gènes ayant déjà été associés aux troubles du neuro-développement), il devient évident que de nombreuses réponses se trouvent également dans les ARN non codants, composants essentiels du splicéosome majeur.
L’élargissement de l’analyse des gènes codant les petits ARN non codants du splicéosome permet désormais d’apporter un diagnostic précis à de nombreux enfants restés sans diagnostic après des tests classiques centrés sur l’ADN codant.
Enfin, l’étude de groupes plus importants de patients, issus de diverses origines ethniques, contribuera à affiner la compréhension des causes moléculaires des troubles du neuro-développement et à évaluer leurs conséquences fonctionnelles. Ces avancées auront un impact concret et significatif pour les familles d’enfants concernés par ces maladies neurogénétiques à travers le monde.
Pour en savoir plus :
Okamoto N, Nishi E, Hasegawa Y, et al. Clinical Study of Nine Patients With ReNU Syndrome. Am J Med Genet A. 2025 Nov ; 197 (11) : e64151. doi : 10.1002/ajmg.a.64151
Di Letto P, De Leonibus C, Palmieri FP, et al ; Telethon Undiagnosed Diseases Program Study Group ; Nigro V, Torella A. Reanalysis of Undiagnosed Neurodevelopmental Disorder Cases : From RNU4-2 Variants to Clinical Phenotypes. Neurol Genet. 2025 Oct 20 ; 11 (6) : e200312. doi : 10.1212/NXG.0000000000200312
Leitão E, Santini A, Cogne B, et al. Systematic analysis of snRNA genes reveals frequent RNU2-2 variants in dominant and recessive developmental and epileptic encephalopathies. medRxiv [Preprint]. 2025 Sep 4 : 2025.09.02.25334923. doi : 10.1101/2025.09.02.25334923
Greene D, Mendez R, Lees J, et al. Biallelic variants in RNU2-2 cause the most prevalent known recessive neurodevelopmental disorder. medRxiv. 2025 [Preprint]. Aug 29 : 2025.08.26.25334179. doi : 10.1101/2025.08.26.25334179
Deutsch HM, Song Y, Li D. Spliceosome complex and neurodevelopmental disorders. Curr Opin Genet Dev. 2025 Aug ; 93 : 102358. doi : 10.1016/j.gde.2025.102358
Rius R, Blakes AJM, Chen Y et al. Biallelic variants in the non-coding RNA gene RNU4-2 cause a recessive neurodevelopmental syndrome with distinct white matter changes. MedRxiv. 2025 [Preprint]. Aug16 : 2025.08.13.25333306
Bertoli-Avella AM, Ganoza CA, Ferreira M, et al. RNU4-2 monoallelic variants as a leading cause of syndromic neurodevelopmental disorder, including in patients with parental consanguinity. J Med Genet. 2025 Jul 21 ; 62 (8) : 536-539. doi : 10.1136/jmg-2024-110556
Jackson A, Thaker N, Blakes A, et al. Analysis of R-loop forming regions identifies RNU2-2 and RNU5B-1 as neurodevelopmental disorder genes. Nat Genet. 2025 Jun ; 57 (6) : 1362-1366. doi : 10.1038/s41588-025-02209-y
Nava C, Cogne B, Santini A, et al. Dominant variants in major spliceosome U4 and U5 small nuclear RNA genes cause neurodevelopmental disorders through splicing disruption. Nat Genet. 2025 Jun ; 57 (6) : 1374-1388. doi : 10.1038/s41588-025-02184-4
Bruselles A, Mancini C, Chiriatti L, et al. Expanding the mutational spectrum of ReNU syndrome : insights into 5’Stem-loop variants. Eur J Hum Genet. 2025 Apr ; 33 (4) : 432-440. doi : 10.1038/s41431-025-01820-1
De Jonghe J, Kim HC, Adedeji A, et al. Saturation genome editing of RNU4-2 reveals distinct dominant and recessive neurodevelopmental disorders. medRxiv [Preprint]. 2025 Apr 10 : 2025.04.08.25325442. doi : 10.1101/2025.04.08.25325442
Barbour K, Friedman J, Bird LM, et al. The Prevalence of RNU4-2-Associated Autosomal Dominant Intellectual Disability Syndrome. Pediatr Neurol. 2025 Mar ; 164 : 1-3. doi : 10.1016/j.pediatrneurol.2024.12.005
Burns VF, Radford EJ. ReNU syndrome – a newly discovered prevalent neurodevelopmental disorder. Trends Genet. 2024 Nov ; 40 (11) : 914-916. doi : 10.1016/j.tig.2024.09.005
Chen Y, Dawes R, Kim HC, et al. De novo variants in the RNU4-2 snRNA cause a frequent
neurodevelopmental syndrome. Nature. 2024 Aug ; 632 (8026) : 832-840. doi : 10.1038/s41586-024-07773-7
Greene D, Thys C, Berry IR, et al. Mutations in the U4 snRNA gene RNU4-2 cause one of the most prevalent monogenic neurodevelopmental disorders. Nat Med. 2024 Aug ; 30 (8) : 2165-2169. doi : 10.1038/s41591-024-03085-5
Griffin C, Saint-Jeannet JP. Spliceosomopathies : Diseases and mechanisms. Dev Dyn. 2020 Sep ; 249 (9) : 1038-1046. doi : 10.1002/dvdy.214
Nguyen TH, Galej WP, Bai XC, et al. The architecture of the spliceosomal U4/U6.U5 tri-snRNP. Nature. 2015 Jul 2 ; 523 (7558) : 47-52. doi : 10.1038/nature14548
Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol. 2014 Feb ; 15 (2) : 108-21. doi : 10.1038/nrm3742
Association française du syndrome de Renu








