



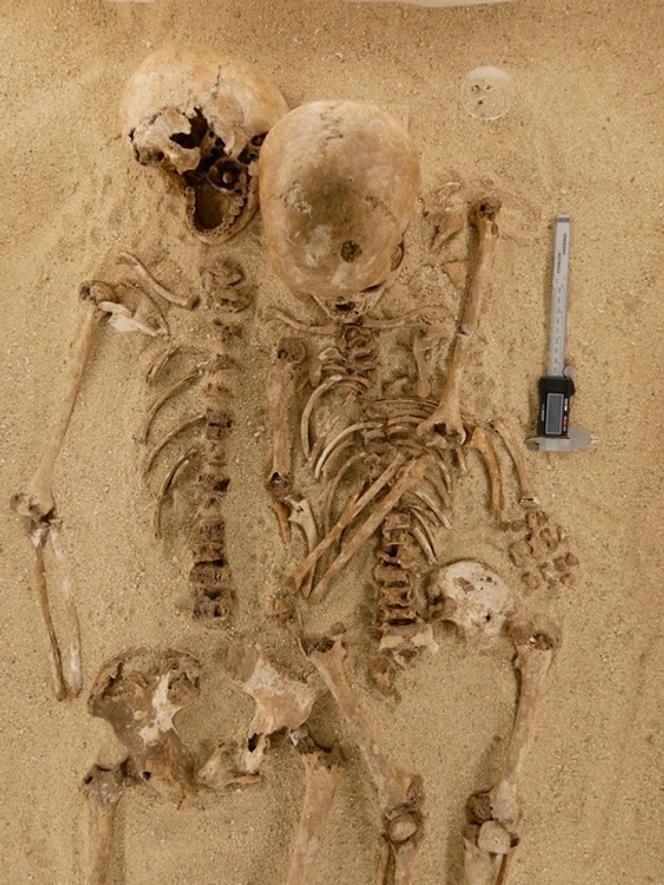
C’est l’histoire d’une personne de petite taille qui a vécu il y a plus de 12 000 ans. Son squelette a été découvert en 1963 dans la grotte de Romito, près de Papasidero (province de Cosenza, région de Calabre, Italie). Elle est morte à l’adolescence. Sa taille est estimée à environ 1,10 m. À ses côtés reposait un autre individu, un adulte mesurant environ 1,45 m. Les deux corps ont été déposés dans la même tombe à la fin du Paléolithique supérieur, dans une position d’étreinte : l’adulte, appelé Romito 1, passait son bras gauche autour de l’adolescente, Romito 2.
Les anthropologues ont observé chez Romito 2 des anomalies osseuses évocatrices d’une maladie rare : la dysplasie acromésomélique de type Maroteaux, affection touchant la croissance et la morphologie du squelette. Il ne s’agissait toutefois que d’une hypothèse fondée sur la seule analyse morphologique des os.
Le terme acromésomélique décrit précisément la distribution des atteintes : « acro » renvoie aux extrémités (mains, pieds), « méso » aux segments intermédiaires des membres (avant-bras, jambes) et « mélie » aux membres eux-mêmes. Dans cette dysplasie, les segments moyens (avant-bras, jambes) et distaux (mains, pieds) sont particulièrement raccourcis, tandis que le tronc est relativement préservé, ce qui entraîne une petite taille disproportionnée.
Le « type Maroteaux », décrit en 1971 par le pédiatre et généticien français Pierre Maroteaux, correspond à un trouble du développement osseux d’origine génétique, caractérisé par une petite taille sévère, un raccourcissement marqué des segments moyens et distaux des membres et un certain degré d’atteinte vertébrale.

Parmi les différentes dysplasies osseuses, cette forme est rare et à transmission autosomique récessive : elle survient lorsqu’une personne hérite de deux copies mutées du même gène, l’une de chaque parent.

La voûte crânienne de Romito 2 est bombée, au-dessus d’une racine nasale peu marquée. Les avant-bras présentent un raccourcissement du radius et du cubitus, avec une incurvation manifeste du radius. Ces anomalies limitaient l’amplitude des mouvements du coude en raison de subluxations ou de luxations.
Au niveau des mains, les métacarpiens sont raccourcis et élargis. Les doigts sont en forme de cône et les phalanges sont courtes. Les pieds montrent également un raccourcissement et un épaississement des métatarsiens et des phalanges, à l’exception du gros orteil, généralement proéminent dans cette affection.
Pendant des décennies, les chercheurs se sont interrogés sur les raisons de cette inhumation conjointe, sur le lien de parenté entre les deux individus et sur l’origine précise des anomalies squelettiques de l’adolescente. Le sexe biologique des deux personnes, leur lien de parenté, leur appartenance à un groupe génétique donné et la nature exacte de la maladie demeuraient inconnus.
Un pont entre archéologie, médecine clinique et génomique
L’analyse de l’ADN ancien, menée plus de soixante ans après la découverte des squelettes, a permis d’apporter des réponses précises. Les résultats ont été publiés le 29 janvier 2026 dans l’hebdomadaire médical américain The New England Journal of Medicine.
Une équipe internationale a entrepris d’extraire de l’ADN ancien à partir des restes osseux datant de la fin du Paléolithique supérieur. Conduite par des chercheurs de l’université de Liège et de Vienne, elle réunissait aussi des biologistes de l’université de Coimbra (Portugal), de l’Institut de recherche environnementale de Wageningen (Pays-Bas) et de l’université Sapienza de Rome.
Analyse de l’ADN de l’os temporal renfermant l’oreille interne
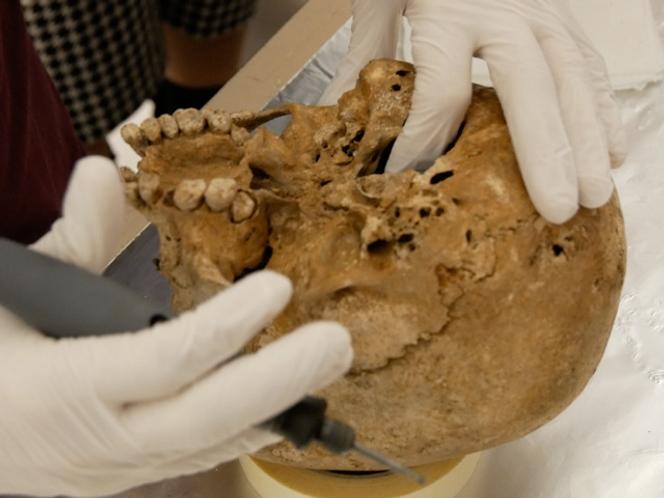
L’ADN ancien est extrait de tissus qui ont traversé des millénaires : il est très fragmenté, chimiquement altéré et souvent présent en quantité infime. Pour maximiser leurs chances d’en récupérer, les chercheurs ont prélevé le matériel génétique au niveau de l’oreille interne gauche de chaque squelette, plus précisément dans la partie pétreuse de l’os temporal. Des travaux antérieurs ont montré que cet os extrêmement dense est l’un des meilleurs sites pour ce type d’analyses. C’est en effet là que l’on obtient des pourcentages exceptionnellement élevés d’ADN ancien d’un individu.
La partie pétreuse de l’os temporal (constituée du rocher) se situe à la base du crâne. Elle abrite les organes de l’audition et de l’équilibre, regroupés dans le labyrinthe osseux comprenant la cochlée, le vestibule et trois canaux semi-circulaires. Il s’agit de la région osseuse la plus dense de tout le corps des mammifères.
Les biologistes moléculaires ont ensuite utilisé des techniques de séquençage à haut débit pour lire les fragments d’ADN et les comparer à des génomes de référence.
Deux femmes apparentées au premier degré
Les analyses ont montré que les deux individus étaient de sexe féminin et apparentées au premier degré : soit une mère et sa fille, soit deux sœurs.
Leur génome a été comparé à un vaste ensemble de génomes modernes et anciens d’Eurasie occidentale, c’est-à-dire des régions d’Europe et de l’ouest de l’Asie. L’étude a situé l’adulte et l’adolescente au sein de ce que les généticiens appellent le « cluster de Villabruna », du nom d’un individu découvert dans le nord de l’Italie (Vénétie) et représentant un groupe de chasseurs-cueilleurs qui, il y a environ 14 000 ans, s’est diffusé à partir du sud de l’Europe vers le centre et l’ouest du continent.
Pour comprendre la pathologie de l’adolescente, l’équipe a séquencé un panel de 36 gènes connus pour être impliqués dans des pathologies squelettiques ou endocriniennes. Ils ont identifié chez Romito 2 une mutation à l’état homozygote dans le gène NPR2, situé sur le chromosome 9.
Mutation ponctuelle dans un gène impliqué dans la croissance osseuse
Ce gène code un récepteur dénommé NPR2 (natriuretic peptide receptor 2), qui joue un rôle central dans la régulation de la croissance osseuse. Ce récepteur est notamment présent dans le cartilage des plaques de croissance, ces zones situées aux extrémités des os longs où l’os s’allonge pendant l’enfance et l’adolescence. Il participe à la formation de l’os à partir du cartilage (l’ossification endochondrale) et à la croissance des os longs en contrôlant la multiplication et la maturation progressive des chondrocytes, les cellules qui fabriquent le cartilage.
Lorsque le gène NPR2 est muté et que le récepteur fonctionne mal, la signalisation est diminuée. Les cartilages de croissance produisent moins d’os en longueur, en particulier dans les segments distaux des membres. C’est le mécanisme moléculaire de la dysplasie acromésomélique de type Maroteaux.
Le plus ancien diagnostic génétique jamais réalisé chez l’humain
L’anomalie identifiée est une mutation ponctuelle de l’ADN (ou variant). On observe la substitution d’une cytosine (C) par une thymine (T) à un emplacement précis dans la séquence codante du gène. La lettre C est ainsi remplacée par la lettre T à la position 1801 (c.1801C→T). Cette modification (« mutation faux-sens ») de l’ADN entraîne le remplacement d’un acide aminé par un autre, en l’occurrence d’une arginine par une cystéine au niveau de la protéine, en position 601 (p. Arg601Cys). Cela intervient dans un domaine au sein duquel certains acides aminés sont extrêmement conservés au cours de l’évolution, c’est-à-dire restés identiques dans de nombreuses espèces au fil du temps, ce qui suggère qu’ils sont cruciaux pour le fonctionnement de la protéine.
La variante du gène NPR2 identifiée chez Romito 2 est extrêmement rare dans les bases de données des populations actuelles. De plus, aucun individu n’y est signalé comme homozygote, c’est-à-dire porteur de deux copies de ce variant particulier. En revanche, des mutations homozygotes affectant ce même résidu ont déjà été décrites chez des patients atteints de dysplasie acromésomélique.
L’adulte Romito 1 ne porte qu’une seule copie mutée du gène NPR2 : elle est ce qu’on appelle une hétérozygote. Il a été montré que des mutations faux-sens du gène NPR2, présentes à l’état homozygote et touchant l’arginine 601, sont responsables de la dysplasie acromésomélique de type Maroteaux. Chez des patients contemporains, des variants hétérozygotes de NPR2, y compris la mutation identifiée chez Romito 2 (responsable de la substitution d’une arginine par une cystéine en position 601), ont par ailleurs été rapportés chez des personnes de petite taille.
L’ensemble de ces données permet donc de poser un diagnostic génétique chez cette adolescente morte au Paléolithique supérieur.

Les chercheurs considèrent cette variante comme probablement pathogène en s’appuyant sur les critères de l’American College of Medical Genetics and Genomics. Par ailleurs, des outils bioinformatiques, qui utilisent différents algorithmes pour prédire l’impact fonctionnel d’un variant sur une protéine, concluent à des effets délétères ou dommageables de cette substitution sur la fonction de la protéine dans plus de 85 % des cas.
L’ensemble de ces éléments plaide donc fortement pour que cette mutation ponctuelle du gène NPR2 joue un rôle déterminant dans le développement de la dysplasie osseuse observée chez Romito 2.
L’étude ne se limite pas à un diagnostic moléculaire rétrospectif. Elle éclaire aussi les conditions de vie d’une adolescente atteinte d’une maladie invalidante dans un groupe de chasseurs-cueilleurs du Paléolithique supérieur. Les anomalies des avant-bras, avec incurvation du radius et limitation des mouvements du coude, devaient entraver les gestes quotidiens. Les mains et les pieds raccourcis et élargis modifiaient la préhension et la marche. Dans un environnement où la mobilité, la chasse et la collecte étaient essentielles, ces limitations pouvaient représenter un handicap réel.
Cette adolescente a pourtant survécu jusqu’à la fin de l’adolescence, avec un régime alimentaire et un niveau de stress nutritionnel comparables à ceux des autres individus du site. Cette observation suggère qu’elle a bénéficié de soins et d’un soutien au sein de son groupe familial.
Des travaux antérieurs, menés notamment par l’archéologue australienne Lorna Tilley, avaient déjà proposé, à propos de Romito 2, une approche de « bioarchéologie du care », autrement dit une réflexion sur les formes d’entraide et de prise en charge des personnes handicapées dans les sociétés préhistoriques.
Le fait qu’une adolescente porteuse d’un handicap congénital sévère ait vécu jusqu’à la fin de l’adolescence et qu’elle ait été inhumée avec un soin manifeste, enlacée avec une proche, renforce l’idée que ces groupes de chasseurs-cueilleurs n’étaient pas exclusivement centrés sur la survie immédiate, mais savaient aussi intégrer des individus « différents », aux capacités physiques très limitées. Tout indique qu’elle a bénéficié de véritables soins et d’une adaptation du groupe à sa différence, une forme d’« accommodation à la différence » avant la lettre.
Les promesses de l’ADN ancien appliqué aux maladies rares
Sur le plan génétique, ce travail démontre qu’il est possible de poser un diagnostic précis sur un individu ayant vécu il y a plus de 12 millénaires. Elle était atteinte de dysplasie acromésomélique de type Maroteaux liée à un variant pathogène du gène NPR2. Il rappelle aussi que les maladies génétiques que nous considérons comme rares aujourd’hui étaient déjà présentes dans les sociétés préhistoriques, composées de groupes humains par ailleurs capables d’adaptation et de solidarité.
Dans le cas de Romito 1 et Romito 2, cette étude sur de l’ADN ancien permet de confirmer un diagnostic posé initialement sur la seule base de la morphologie squelettique, de préciser l’anomalie génétique sous-jacente, de documenter la transmission familiale de la maladie et de replacer cette histoire individuelle dans le contexte plus large des populations préhistoriques européennes.
Ce travail montre aussi qu’il est désormais possible de cibler des ensembles précis de gènes impliqués dans des maladies et de les analyser dans des restes humains vieux de plus de dix mille ans, ouvrant la voie à une véritable paléogénétique clinique. Cette découverte marque donc une étape importante pour la compréhension de l’histoire des maladies rares.
« Ces deux personnes qui ont vécu au Paléolithique ont été courageuses et ont fait de leur mieux dans des circonstances incroyablement difficiles », me confie Adrian Daly, endocrinologue au CHU de Liège et coresponsable avec Ron Pinhasi, anthropologue évolutionniste à l’université de Vienne, de l’étude parue dans le NEJM.
Selon ces auteurs, « cette approche, qui permet de poser un diagnostic génétique à partir de restes humains préhistoriques, pourrait être utilisée pour mieux évaluer la charge que représentent les maladies génétiques rares au sein des populations humaines anciennes ».
Que retenir de cette étude ? D’abord qu’il est désormais possible d’identifier, à partir d’ADN ancien, la cause moléculaire précise d’une maladie observée sur des os vieux de plus de dix millénaires. Ensuite que les maladies génétiques dites « rares » ne sont pas des anomalies récentes : elles traversent l’histoire humaine depuis des milliers d’années. Enfin, ce cold case paléolithique ne raconte pas seulement l’histoire d’une jeune patiente porteuse d’une mutation. Il montre que l’archéologie et la médecine peuvent désormais dialoguer à l’échelle du génome.
Pour en savoir plus :
Fernandes DM, Llanos-Lizcano A, Brück F, et al. A 12,000-Year-Old Case of NPR2-Related Acromesomelic Dysplasia. N Engl J Med. 2026 Jan 29 ; 394 (5) : 513-515. doi : 10.1056/NEJMc2513616
Jeong R, Covarrubias S, Shanghavi D, et al. Functional analysis of NPR2 variants supports the therapeutic rationale for CNP in short stature. Am J Hum Genet. 2026 Jan 8 ;113(1) :117-132. doi : 10.1016/j.ajhg.2025.12.006.
Badawi S, Varghese DS, Raj A, John A, et al. Unveiling the pathogenic mechanisms of NPR2 missense variants : insights into the genotype-associated severity in acromesomelic dysplasia and short stature. Front Cell Dev Biol. 2023 Nov 23 ; 11 : 1294748. doi : 10.3389/fcell.2023.1294748
Coqueugniot H. L’enfant « différent » au Paléolithique. Les Nouvelles de l’archéologie, 2021, 165, pp.38-43. doi : 10.4000/nda.12873. halshs-03799372
Irfanullah, Umair M, Khan S, Ahmad W. Homozygous sequence variants in the NPR2 gene underlying Acromesomelic dysplasia Maroteaux type (AMDM) in consanguineous families. Ann Hum Genet. 2015 Jul ; 79 (4) : 238-44. doi : 10.1111/ahg.12116
Pinhasi R, Fernandes D, Sirak K, et al. Optimal Ancient DNA Yields from the Inner Ear Part of the Human Petrous Bone. PLoS One. 2015 Jun 18 ; 10 (6) : e0129102. doi : 10.1371/journal.pone.0129102
Mallegni F, Fabbri PF. The human skeletal remains from the Upper Palaeolithic burials found in Romito cave (Papasidero, Cosenza, Italy). Bull Mem Soc Anthropol Paris 1995 ; 7 : 99-137.
Frayer DW, Horton WA, Macchiarelli R, Mussi M. Dwarfism in an adolescent from the Italian late Upper Palaeolithic. Nature. 1987 Nov 5-11 ; 330 (6143) : 60-2. doi : 10.1038/330060a0
Maroteaux P, Martinelli B, Campailla E. Le nanisme acromésomélique. Presse Med (1893). 1971 Oct 9 ; 79 (42) : 1839-42. PMID : 5000841.
N.B. : Je remercie chaleureusement le Dr Adrian Daly de m’avoir communiqué les photographies qu’il a réalisées au Museo Archeologico Nazionale di Reggio Calabria.








